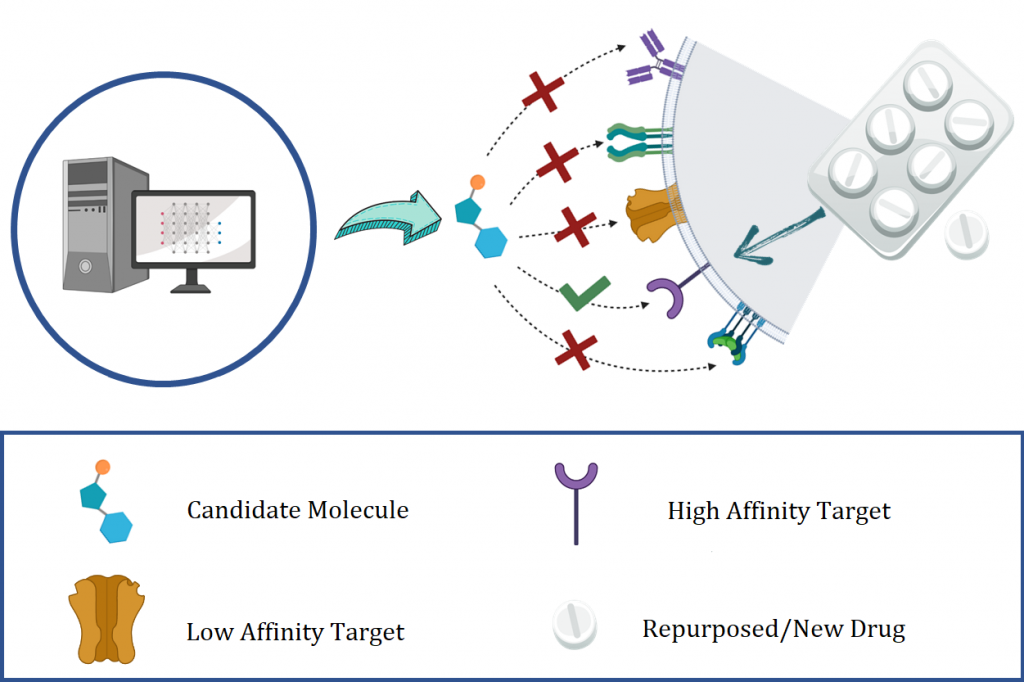
Drug Discovery is an essential endeavor to tackle threats to human health. Notwithstanding, the development and subsequent market penetration of new pharmaceuticals is a critical yet time-consuming and expensive process. To address shortcomings in this process, new approaches have been explored to combine both experimental and computational routes. In particular, as an in-silico approach, virtual screening is proposed as an alternative to identify active molecules towards therapeutic biological targets. Recent advances in artificial intelligence (AI) have provided more effective search algorithms while reducing the time it takes to do virtual screening and implementing new therapeutic candidates.
In this research line, we employ different state-of-the-art techniques of deep learning (specifically Natural Language Processing (NLP), Artificial Vision, and Graph Networks) to model relationships between molecules, proteins, and the variables involved in the task of predicting the affinities among ligands and targets.