Skip to main content
Home
People
Research
Global Health
Computer Vision
Sustainability
Publications
PHARMANET: PHARMACEUTICAL DISCOVERY WITH DEEP RECURRENT NEURAL NETWORKS
Home
Research Publications
PHARMANET: PHARMACEUTICAL DISCOVERY WITH DEEP RECURRENT NEURAL NETWORKS
Abstract
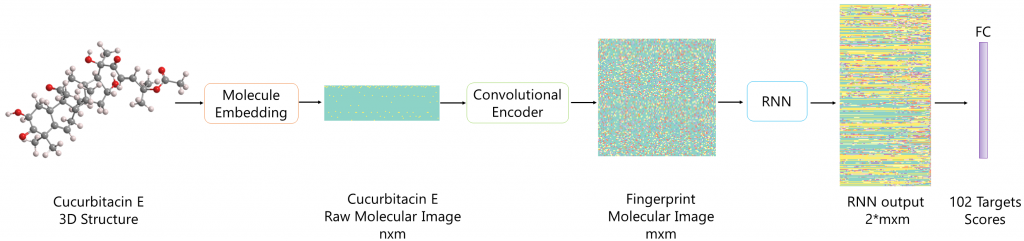
Method
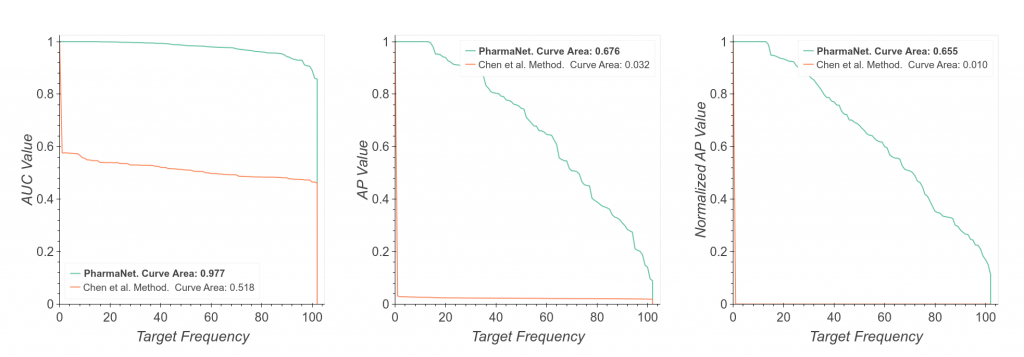
Results